Autores: Juliana G. Freitas, Miguel A. A. Soto, Elias H. Teramoto, Veronica Gonsalez e Chang H. Kiang
1. Partição e fases dos contaminantes
O meio subterrâneo é composto pela fase sólida e pelos poros, que podem conter diferentes fluidos (como água e ar). A fase sólida inclui partículas minerais e matéria orgânica de solos, sedimentos e rochas. Em áreas contaminadas, os contaminantes podem se distribuir entre os sólidos e os fluidos, estando em diferentes fases. Até três fluidos podem estar presentes nos poros: a fase gasosa, a fase aquosa e uma fase líquida não aquosa, também chamada de NAPL (non-aqueous phase liquid). Os contaminantes também podem estar retidos nos sólidos, denominada de fase adsorvida ou sorvida.
Cada fase pode ser composta por uma variedade de compostos. A fase aquosa, por exemplo, é composta principalmente de água, mas contém compostos orgânicos e inorgânicos dissolvidos. Ao contaminante presente na fase aquosa, dá-se o nome de fase dissolvida. Os contaminantes se distribuem entre essas diferentes fases dependendo das suas propriedades e das condições do meio. A transferência de massa entre fases (partição) ocorre até que sejam atingidas condições de equilíbrio químico.
1.1 Dissolução / Precipitação
Os processos de dissolução e precipitação se referem à partição entre a fase aquosa e o contaminante em fase separada, podendo ser como NAPL ou um sólido. A propriedade que descreve essa partição é a solubilidade (S), que expressa a concentração da substância em fase dissolvida quando em equilíbrio com a sua fase pura (líquida ou sólida). Se a fase separada não é um composto puro, mas uma mistura de substâncias, o equilíbrio químico multicomponente é mais complexo e, de forma geral, dependente da fração molar do composto na mistura.
A solubilidade varia ligeiramente com a temperatura, mas também pode variar dependendo da presença de outras substâncias em solução e das condições físico-químicas. Para compostos orgânicos, destaca-se que substâncias conhecidas surfactantes ou cossolventes (como etanol) podem causar um grande aumento da solubilidade. No caso de algumas substâncias inorgânicas, as condições físico-químicas (como pH e potencial redox) da solução têm um grande impacto na solubilidade.
Quando a concentração na fase dissolvida é inferior ao que seria esperado em condições de equilíbrio, o contaminante presente em fase separada vai se dissolver até que seja atingido o equilíbrio, ou até a fase separada se extinguir.
1.2 Volatilização
O contaminante presente como fase dissolvida ou fase separada (NAPL ou sólido) pode particionar para a fase gasosa, pelo processo conhecido como volatilização. Esse processo acontece comumente com compostos orgânicos na zona não saturada ou saturada.
A tendência de volatilização de um contaminante pode ser avaliada por duas propriedades: a pressão de vapor e a constante de Henry. A pressão de vapor, que indica a pressão do contaminante em equilíbrio com sua fase pura, líquida ou sólida, é uma medida da tendência de evaporação dessa substância.
Em misturas de compostos orgânicos (como no caso da gasolina), a pressão de vapor de uma substância na mistura será menor que da substância pura, e pode ser aproximada pela Lei de Raoult, que determina que a pressão de vapor da substância na mistura é o produto da pressão de vapor da substância pura e da fração molar do composto na mistura.
A segunda propriedade é a constante da Lei de Henry, que descreve a partição entre a fase dissolvida e a fase vapor. A Lei de Henry considera que existe uma relação linear entre as concentrações de equilíbrio na fase vapor e em uma solução aquosa diluída. A constante de Henry é então definida como a razão entre a concentração na fase vapor e na fase líquida. Portanto, quanto maior a constante de Henry mais volátil é o composto.
1.3 Sorção
Sorção é definida como a interação de um contaminante com um sólido (como solo, sedimento ou matriz rochosa), sendo dividida em adsorção e absorção. De forma geral, adsorção se refere a um acúmulo do contaminante na superfície do sólido, enquanto absorção implica na penetração do contaminante na fase sólida, de forma relativamente uniforme. Uma série de processos pode resultar em sorção, como reações químicas nas superfícies (hidrólise, complexação, troca iônica, formação de pontes de hidrogênio), interações eletrostáticas e interações hidrofóbicas.
A sorção depende das propriedades físicas e químicas do contaminante, da composição da fase sólida e das propriedades do fluido em que se encontra o contaminante e o sólido. Na fase sólida, destaca-se a importância da fração argila (grãos <0,002 mm), incluindo minerais e matéria orgânica, como os principais sólidos para a sorção de contaminantes. A maior sorção nessa fração está associada à elevada área superficial específica e ocorrência de cargas elétricas. A maior parte dos argilominerais apresenta carga elétrica negativa, que favorece a adsorção de cátions. Além disso, minerais da fração argila também podem apresentar carga variável, dependente do pH. As cargas variáveis são especialmente importantes para os óxidos, hidróxidos e óxi-hidróxidos, podendo gerar cargas positivas significativas. O carbono orgânico também desempenha papel importante para a sorção, principalmente, para os contaminantes orgânicos sintéticos. As substâncias iônicas apresentam comportamento complexo relativo à sorção, dependente das condições de pH.
A sorção de contaminantes interfere no transporte dos contaminantes no meio subterrâneo. Assim, o coeficiente de partição, ou coeficiente de distribuição (Kp ou Kd), é um dos parâmetros mais importantes para entender o transporte de poluentes. Esse coeficiente indica a distribuição de uma substância entre a fase sólida e a fase líquida, considerando condições de equilíbrio, e pode variar muitas ordens de grandeza dependendo da substância e do meio.
Ressalta-se que a sorção é um processo complexo, muitas vezes dependente de vários fatores, como pH, força iônica, proporção sólidos-água e outras substâncias dissolvidas. Assim, valores de Kd ou Kp estimados a partir de outras propriedades (como do coeficiente de partição octanol-água: Kow) ou até mesmo determinados em laboratório devem ser considerados como uma aproximação do comportamento do contaminante no meio.
2. Comportamento de fases líquidas não aquosas – NAPLs
Líquidos orgânicos imiscíveis com a água podem estar presentes em uma área contaminada como uma fase oleosa separada da água, denominada de fase líquida não aquosa ou NAPL (non-aqueous phase liquids). Essas fases oleosas podem ser compostas por um único composto ou ser uma mistura de vários constituintes com diferentes propriedades, como é o caso da gasolina, uma mistura de centenas de compostos. No entanto, NAPLs não são completamente insolúveis em água, e podem se dissolver em taxas lentas, resultando na contaminação da água subterrânea. Além disso, alguns compostos podem volatilizar contribuindo para geração da fase vapor. O transporte e o comportamento dos NAPLs em subsuperfície são controlados pela gravidade, forças viscosas e capilares. Entre as propriedades que controlam seu comportamento destaca-se a densidade e, baseado nisso, os NAPLs são classificados em duas categorias: i) LNAPLs, que apresentam densidade inferior à da água (como gasolina e óleo diesel), e; ii) DNAPLs, com densidade superior à da água (como os solventes clorados tetracloroeteno, tricloroeteno e tricloroetano). No caso de NAPLs que são uma mistura de compostos, a densidade pode ser estimada pela média das densidades de cada composto ponderada pela fração volumétrica do composto na mistura. A Figura 1 mostra alguns NAPLs de diferentes composições.
O NAPL pode estar presente como fase livre ou fase residual. A diferença entre as duas fases é a mobilidade. A fase livre é móvel, podendo migrar para dentro de poços de monitoramento, por exemplo. Isso ocorre quando o volume do NAPL em relação ao volume de poros (saturação de NAPL) é suficiente para existir continuidade do produto nos poros. A fase residual é imóvel, não apresenta continuidade, estando retida por forças capilares. Na zona não saturada, em geral, a fase residual está entre a água e o ar, na forma de filmes e anéis (Figura 2). Na zona saturada, a fase residual pode ter formato de bolhas, que podem ser formadas, por exemplo, quando a água ocupa um espaço previamente ocupado pelo NAPL, e separa e isola uma parte do produto do corpo do NAPL. A mobilidade do NAPL pode ser descrita pela permeabilidade relativa, que é uma função da saturação do produto. Esse parâmetro varia de zero a um, sendo que quando a saturação é 100% (todos os poros estão preenchidos por NAPL) a permeabilidade relativa é 1. Quando a saturação é igual à saturação residual (todo o NAPL está como fase residual) a permeabilidade relativa é zero.
2.1 Migração de LNAPLs
Quando ocorre um vazamento de LNAPL, sua migração na zona não saturada ocorre predominantemente na direção vertical, em fluxo descendente. O espalhamento lateral pode ocorrer pelo efeito de forças capilares e pela presença de heterogeneidades na litologia. Durante a migração nessa zona, o LNAPL desloca principalmente o ar (que possui maior mobilidade), mas uma pequena parcela de água também pode ser deslocada. Ao longo da sua migração, parte do NAPL fica retida como fase residual (Figura 2). Se o volume de produto for pequeno, a migração poderá cessar na zona não saturada, com todo o volume retido na fase residual.
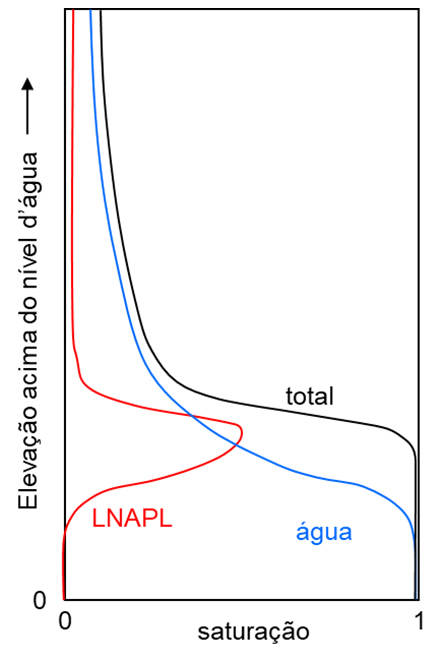
Se o volume de produto vazado for maior, o LNAPL continua avançando em direção à zona saturada. À medida que o LNAPL atinge maiores profundidades, aumenta a saturação de água e a mobilidade do LNAPL diminui devido ao compartilhamento do espaço dos poros entre os dois fluidos (diminuição da permeabilidade relativa). Combinado a isso, a sua menor densidade em relação à água gera forças empuxo que fazem com que o LNAPL tenda a se acumular na zona da franja capilar, causando um espalhamento lateral. A Figura 3 ilustra um perfil típico de saturações (volume de poros ocupado pelo fluido) resultante de um vazamento de LNAPL. Se o volume do vazamento for grande o suficiente, a pressão de LNAPL aumenta e ele pode penetrar o nível d’água (NA), além de gerar um maior espalhamento lateral. Quando o fluxo descendente de NAPL cessa devido à exaustão de produto, o NAPL que atingiu profundidades abaixo do NA é deslocado pela água, mas uma fase residual permanece na zona saturada. Oscilações na profundidade do nível d’água podem causar mobilização de parte do LNAPL, levando à formação de fase residual na zona de oscilação.
2.2 Migração de DNAPLs
A migração de DNAPLs na zona não saturada ocorre de forma similar ao descrito para os LNAPLs. Devido à maior densidade a migração lateral pode ser reduzida e o caminho seguido ligeiramente diferente, mas o comportamento é semelhante, com fluxo predominantemente vertical descendente. No entanto, ao atingir o NA, se o volume de DNAPL é suficiente, o DNAPL continua migrando verticalmente. A fase residual de DNAPL também se forma nas zonas não saturada e saturada, por onde o produto migrar.
O principal fator que vai controlar a migração de DNAPL é a presença de materiais de menor permeabilidade, que pode interromper ou desviar o fluxo do DNAPL. Dois mecanismos podem causar o acúmulo de DNAPL em camadas de menor permeabilidade. O primeiro é que o DNAPL precisa ter pressão suficiente para superar as forças capilares no material de menor permeabilidade (mecanismo de barreira capilar). O segundo é a maior resistência ao fluxo na camada menos permeável. Em meios fraturados, para que o DNAPL consiga penetrar uma fratura precisa ter uma pressão suficiente para vencer a pressão capilar na fratura. Essa pressão necessária depende das propriedades do DNAPL (como densidade e tensão interfacial) e da fratura (como tamanho da abertura). A migração de um DNAPL em meio poroso é ilustrada na Figura 2.
Figura 1 Exemplos de NAPLs de diferentes densidades em frascos com água: a) gasolina (0,75 g cm-3); b) tricloroeteno (TCE) (1,46 g cm-3); c) Gasolina + TCE – proporção 2:1 (» 0,9 g cm-3); d) Gasolina + TCE – proporção 1:2 (» 1,2 g cm-3)

Figura 2 Migração de LNAPL e DNAPL em meios porosos e detalhe da fase residual nas zonas não-saturada e saturada
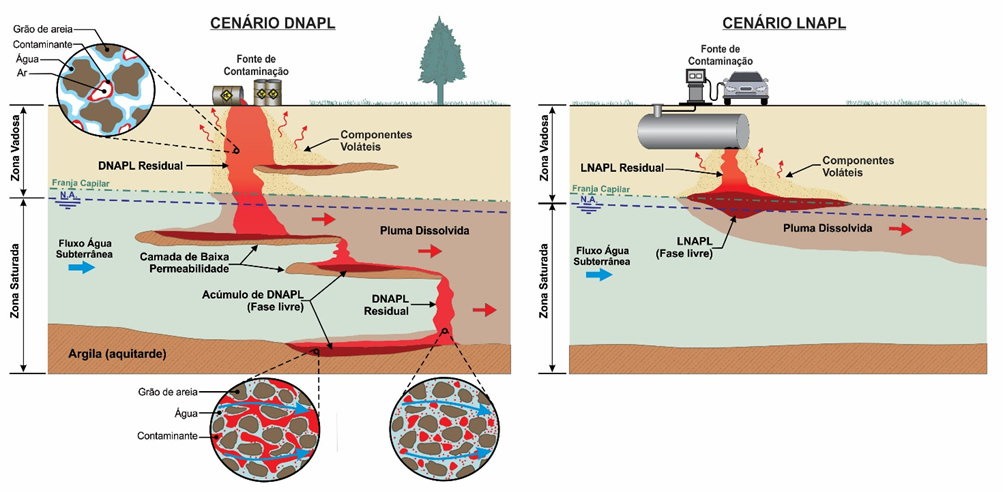
Figura 3 Perfis típicos de saturação de água, LNAPL e total (água + NAPL) num meio homogêneo
3. Transporte de plumas dissolvidas
3.1 Advecção e dispersão
O transporte de substâncias dissolvidas é determinado por dois processos básicos: a advecção e a dispersão. A advecção corresponde ao transporte da substância pelo fluido no qual ela se encontra, na mesma direção e sentido do fluxo do fluido, com velocidade igual à velocidade média do fluido. Assim, um contaminante dissolvido na água subterrânea é transportado por advecção com uma velocidade igual à velocidade média da água subterrânea, como definido no Item 1.4.
A dispersão hidrodinâmica, ou somente dispersão, corresponde a todos os processos que causam espalhamento dos contaminantes ao redor do centro de massa da pluma. Assim, a dispersão causa mistura com água subterrânea não contaminada, causando diluição e expansão da pluma de contaminação para zonas além das que seriam impactadas baseado somente na advecção. A dispersão pode ocorrer na direção do fluxo do fluido, chamada de dispersão longitudinal, ou normal à direção do fluxo, sendo denominada de dispersão lateral ou transversal. A dispersão é causada por processos macroscópicos e microscópicos, e é uma somatória dos processos de difusão molecular e dispersão mecânica.
A difusão molecular é causada pelo movimento aleatório das moléculas, que gera o transporte de soluto da região de maior concentração para as regiões de menor concentração. Assim, a difusão molecular independe do fluxo do fluido, podendo ser o processo dominante em situações em que o gradiente hidráulico é zero ou a condutividade hidráulica muito baixa. Por exemplo, uma situação em que a difusão molecular adquire grande importância é no transporte em meios fraturados onde a matriz rochosa possui baixa condutividade hidráulica de tal forma que o fluxo advectivo é insignificante. Nesses casos, o transporte nas fraturas ocorre por advecção e dispersão, mas parte da massa pode migrar para a matriz rochosa por difusão molecular devido ao gradiente de concentração que se estabelece.
O processo de difusão molecular é descrito pela Lei de Fick, que determina que o fluxo de um soluto (F) é proporcional ao gradiente de concentração (dC/dx), como indicado na Equação 1, para uma dimensão. Em meios porosos a difusão ocorre mais lentamente que na água, pois o transporte somente ocorre pelos poros, resultando em colisões das moléculas com os sólidos e percursos mais longos. Para considerar esse efeito utiliza-se o coeficiente de difusão molecular efetivo (D*), que se relaciona ao coeficiente de difusão pela multiplicação por um coeficiente empírico, usualmente estimado com base na porosidade e tortuosidade do meio, ou determinado experimentalmente.
Onde: F: fluxo do soluto [M L-2 T-1]; D* coeficiente de difusão molecular efetivo [L² T-1]; C: concentração [M L-3]; : gradiente de concentração na direção x.
A dispersão mecânica ocorre devido a variações locais de velocidade, que são desvios em relação à velocidade média do fluxo de água. Essas variações podem ocorrer, por exemplo, dentro de um poro, pois no centro do poro a velocidade é maior que próximo à superfície dos grãos. A diferença no tamanho dos poros também causa gradientes de velocidades, pois a velocidade tende a ser maior nos percursos por poros maiores. Além disso, o contaminante pode seguir diferentes percursos, alguns apresentando maiores comprimentos pela presença de maior tortuosidade, ou ainda causando ramificações laterais. Heterogeneidades do aquífero também podem causar dispersão mecânica, quando a pluma atinge regiões com diferentes condutividades hidráulicas, por exemplo. Assim, a dispersão tende a ser maior quanto maior a extensão da pluma, sendo dependente da escala
.A dispersão mecânica é expressa de forma similar à difusão molecular, empregando o coeficiente de dispersão mecânica, Dm. Esse coeficiente é proporcional à velocidade média de fluxo da água subterrânea e à dispersividade, um coeficiente dependente das propriedades do meio. A Equação 2 representa o cálculo do coeficiente de dispersão mecânica em uma direção. De forma geral, a dispersão na direção longitudinal é maior que nas direções perpendiculares ao fluxo de água subterrânea (diferença de pelo menos uma ordem de magnitude), gerando um maior espalhamento nessa direção.
![]()
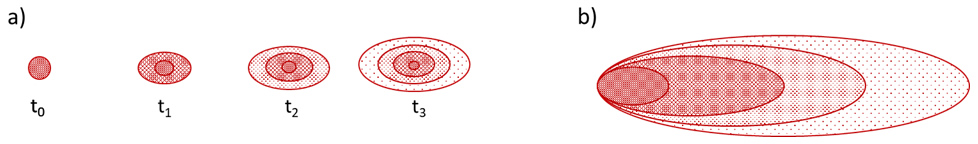
Onde: coeficiente de dispersão mecânica [L2 T-1]; dispersividade na direção x [L]; : velocidade média da água subterrânea na direção x [L T-1].Como a dispersão hidrodinâmica é a somatória dos processos de difusão molecular e dispersão mecânica, define-se o coeficiente de dispersão hidrodinâmica como a soma do coeficiente de difusão molecular efetivo e do coeficiente de dispersão mecânica (Eq. 3). O processo de advecção e dispersão ocorrem conjuntamente, fazendo com que o centro de massa da pluma de contaminação se mova com velocidade igual a velocidade média do fluxo de água subterrânea, e os solutos se espalhem ao redor desse ponto. A junção desses processos é descrita pela equação de advecção-dispersão (Eq. 4), ilustrada na Figura 4. Ressalta-se que heterogeneidades no meio causam desvios no fluxo, gerando plumas de contaminação com geometrias distintas do que seria observado num meio homogêneo.

Figura 4 Exemplo da migração de plumas de contaminação a partir de (a) uma fonte tipo pulso, instantânea e b) uma fonte contínua. Maior densidade de pontos representa maior concentração na fase dissolvida
3.2 Retardamento
A sorção de contaminantes dissolvidos nos sólidos resulta em uma redução na velocidade de transporte das substâncias dissolvidas, mas não na remoção de massa. Assumindo equilíbrio na sorção e comportamento linear (Kd – coeficiente de distribuição – não varia significativamente com a concentração da substância), a velocidade da pluma será menor do que a velocidade média da água subterrânea e é estimada utilizando o fator de retardamento (R). O fator de retardamento é a razão entre a velocidade do centro de massa da pluma do contaminante e a velocidade média de fluxo da água subterrânea e pode ser estimado pela Equação 5.

Onde: vi: velocidade de migração do poluente [L T-1]; : velocidade média da água subterrânea [L T-1]; R: fator de retardamento [-]; : densidade aparente do meio [M L-3]; : coeficiente de partição [L3 M-1]; : porosidade do meio [-]Ressalta-se que essa simplificação em geral é aplicável para compostos orgânicos hidrofóbicos. Para substâncias iônicas, como metais, esse modelo não gera uma boa aproximação, pois a sorção varia muito com as propriedades do meio, como pH, força iônica etc.
| A equação que governa o transporte de substâncias dissolvidas é derivada da lei de conservação de massa considerando os processos de advecção, dispersão mecânica, difusão molecular, transformações bioquímicas (representadas pelo coeficiente de decaimento de 1ª ordem l) e transferência de massa entre fases (sorção). Assim, o transporte unidirecional de solutos pode ser expresso pela equação abaixo: |
4. Transporte da fase vapor
O transporte da fase vapor pode gerar riscos significativos principalmente pela intrusão de vapores, que consiste em um processo de migração de determinados compostos químicos orgânicos e/ou inorgânicos voláteis desde uma contaminação subsuperficial até o interior de construções sobrejacentes.
4.1 Modelo conceitual
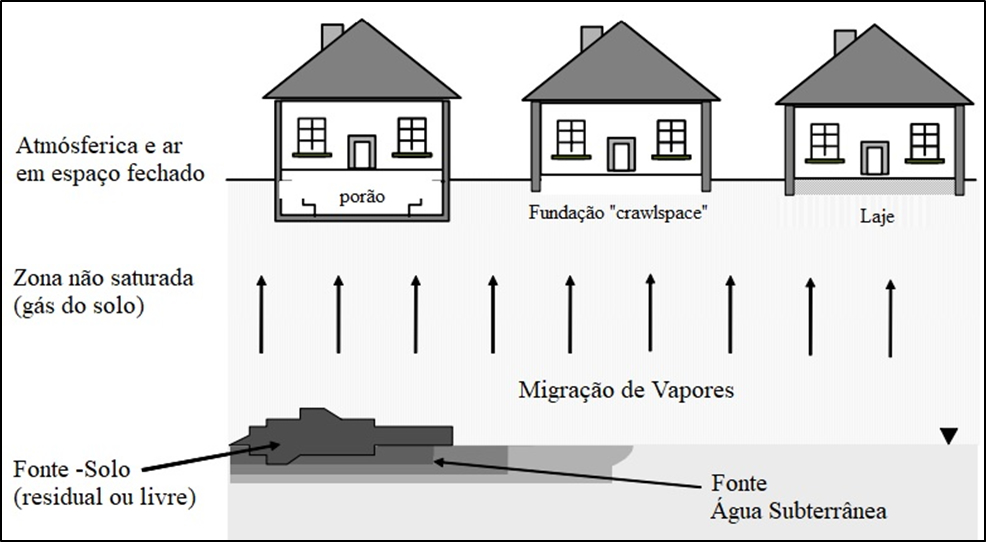
A Figura 5 ilustra o modelo conceitual padrão para explicar a intrusão de vapor proveniente de solo ou água subterrânea contaminada. O modelo conceitual conta com uma Rota de Exposição que compreende o percurso desde a origem da contaminação até o destino final, sendo constituída por: fonte de contaminação secundária, meios físicos, mecanismos de transporte, ponto de exposição, vias de exposição e receptores atuais e futuros.
Figura 5. Intrusão de vapor proveniente de água subterrânea (Adaptado de ITRC, 2014)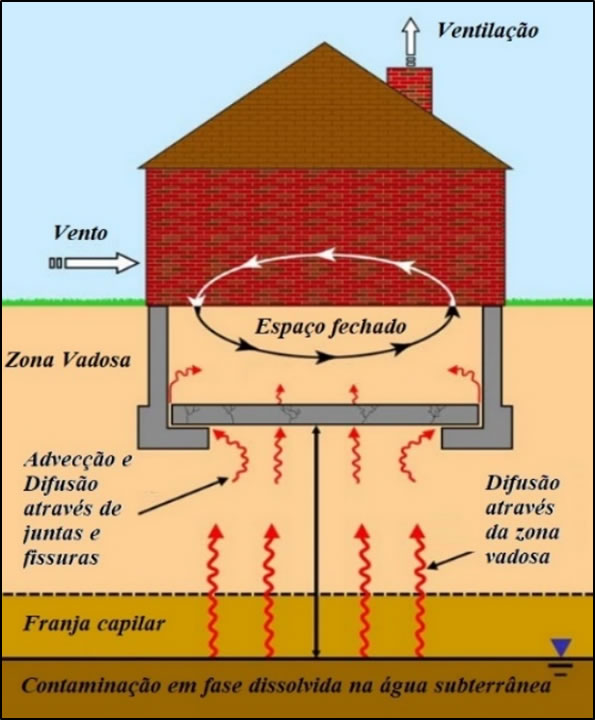
4.1.1 Fonte de contaminação secundária
A fonte de vapores é emanada do solo ou da água subterrânea contaminada com produtos químicos voláteis em concentrações elevadas, cuja magnitude é capaz de oferecer riscos imediatos à segurança (potencial de incêndio ou explosão de vapores de petróleo ou metano) ou possíveis efeitos adversos à saúde humana, pela inalação a longo prazo em ambientes internos sobrejacentes.
Substâncias voláteis são consideradas produtos químicos com constante da lei de Henry maior que 10‑5 atm m3 mol-1 à temperatura ambiente, ou com pressão de vapor maior que 1 mm Hg à temperatura ambiente. Entre os produtos químicos formadores de vapor podem ser citados os mais comuns: hidrocarbonetos encontrados na gasolina, diesel e combustível de aviação (e.g., benzeno, trimetilbenzenos, naftaleno); produtos químicos voláteis de aditivos de combustível (e.g., álcoois, éter metil terciário-butílico, álcool terciário-butílico, dibrometo de etileno e 1,2-dicloroetano); metano, produzido pela biodegradação anaeróbica de contaminantes orgânicos e matéria orgânica no solo, bem como compostos halogenados (e.g., cloreto de vinila, 1,2-dicloroetano e hexaclorobenzeno).
4.1.2 Meio físico
O levantamento detalhado de dados sobre o meio físico, incluindo características geológicas, físicas, físico-químicas, mecânicas e hidrodinâmicas, permite compreender o trajeto dos contaminantes na fase vapor. Adicionalmente, devido a sua interação com a atmosfera, são necessários dados hidrológicos e meteorológicos.
Ainda, a migração de contaminantes em forma de vapor pode ser impedida ou facilitada por diversos fatores, tais como: espessura da zona não saturada, trincas e fraturas, grau de saturação, densidade do vapor, condutividade hidráulica de solo não saturado, permeabilidade ao vapor, granulometria, curva de retenção de água no solo, entre outros. Por exemplo, solos podem apresentar maiores graus de saturação e inibir a magnitude do transporte difusivo, devido ao coeficiente de difusão dos compostos químicos, formadores do vapor, ser substancialmente menor na água em comparação com o ar. A variabilidade do grau de saturação da zona não saturada está sujeita não somente à precipitação e infiltração (às vezes impermeabilizados superficialmente por fundações), mas também às flutuações no nível da água subterrânea.Solos ou camadas de baixa permeabilidade na zona não saturada também podem restringir a migração ascendente de vapores, sendo o efeito acentuado com o aumento de umidade e profundidade da zona contaminada. Além disso, alguns compostos (e.g., benzeno, metano, cloreto de vinila) podem apresentar reduções em suas concentrações de gás no solo devido à biodegradação na zona vadosa.
4.1.3 Mecanismos de transporte da fase vapor
Levando em consideração o modelo conceitual padrão da Figura 5 podem ser identificados três processos de transporte de vapor após a volatilização de substâncias químicas a partir do solo ou água subterrânea: i) difusão na zona não saturada, desde a fonte até o solo adjacente à fundação do edifício; ii) advecção e difusão através de fissuras, trincas e juntas entre a laje e muros da construção; e iii) mistura com o ar interno no ponto de exposição.
Difusão – já definida anteriormente, consiste no movimento de substâncias químicas das áreas de maior concentração para as de menor concentração. A magnitude do fluxo difusivo guarda relação direta com o gradiente de concentração, com o coeficiente de difusão molecular efetivo do composto no meio poroso e com a umidade da zona não saturada. O coeficiente de difusão efetiva em um meio poroso, de acordo com Nielsen et al. (1972), pode ser expresso pela Equação 7. Nessa equação é considerado o conteúdo volumétrico de ar, pois, em um solo não saturado, o espaço poroso é composto por uma parcela de ar e outra de água que se complementam. Além disso, é considerada a tortuosidade do meio. Algumas expressões são apresentadas na literatura para estimar a tortuosidade, considerando parâmetros como a porosidade e o conteúdo volumétrico de água.
![]()
Onde Deff representa a difusão efetiva [L2 T-1]; qa, o conteúdo volumétrico de ar disponível no meio poroso [-]; t, a tortuosidade da fase do ar que é responsável pela diminuição da área da seção transversal e aumento do comprimento do caminho induzido por um meio poroso [-]; e Da, a difusividade no ar [L2 T-1].Caso a fonte de contaminação esteja localizada na água subterrânea é necessário levar em conta a difusão efetiva desde a franja capilar (como uma camada adicional). Nesse caso, a difusão efetiva pode ser determinada com a seguinte expressão:
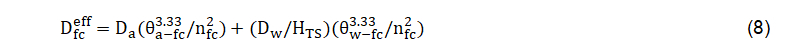
Onde representa a difusão efetiva na franja capilar [L2 T-1]; é a porosidade total na franja capilar e equivalente à umidade volumétrica de saturação [-]; o conteúdo volumétrico de água na franja capilar, correspondente à pressão de entrada de ar que é aproximadamente o inverso do parâmetro α da curva de retenção de água no solo pelo ajuste com o modelo de van Genuchten [-]; e , que representa o conteúdo volumétrico de ar na franja capilar, e que resulta da diferença entre a porosidade total e o conteúdo volumétrico de água na franja capilar [-]; Dw, a difusividade da água [L2 T-1]; HTS, a constante de Henry para a temperatura do sistema [-].
Logo, o coeficiente de difusão efetivo total, levando em consideração o total de camadas de solo, é representado como:

Onde representa o coeficiente global de difusão efetiva da fase vapor de todas as camadas de solo [L2 T-1]; LT, a distância vertical compreendida entre a face inferior da fundação ou laje (adjacente ao ambiente interno) e a fonte de vapores [L]; n, o número de camadas de solo; Li, espessura da camada de solo i [L]; e a difusão efetiva através da camada de solo i [L2 T-1].
Advecção – movimento das substâncias químicas ocorre com o movimento da massa do gás do solo devido a diferenças de pressão. As diferenças de pressão podem ser geradas por mudanças de pressão atmosférica, mudanças de temperatura ou devido a sistemas de ventilação de edifícios, criando convecção natural no solo, isto é, essencialmente fluxo advectivo vertical, conforme o modelo de Johnson e Ettinger (1991), que por ser um modelo unidimensional, conta apenas com a componente vertical e a inexistência da difusão. Cabe ressaltar que esse tipo de transporte é o processo mais significativo nas proximidades da fundação, sendo menos importante distante dela.
Mistura – Os vapores intrudidos podem ser misturados e diluídos com o ar dentro da edificação, a depender da taxa de ventilação. Esses valores podem ser ainda facilitados pelo grau de ventilação que possua o prédio, e.g., ar-condicionado e aquecedores.
4.1.4 Pontos de exposição
O contato dos receptores com as substâncias químicas em forma de vapor poderá ocorrer em diversos tipos de espaços internos ou pontos de exposição, tais como: residências, comércio, escolas, igrejas etc. O tipo de uso, características construtivas e geométricas da edificação e qualquer outra característica que envolva algum tipo de interação com os meios contaminados (temperatura, diferencial de pressão com o solo, distância com a fonte etc.) influenciarão nas magnitudes das doses de ingresso na população afetada nos pontos de exposição.
De acordo com API (2005), a migração do gás do solo e intrusão de vapores pode ser representada por três diferentes cenários construtivos, que possuem desde maior contato (e.g., porão) a menor contato com a superfície do solo (e.g., laje), tal como mostrado na Figura 6. Nessas circunstâncias, levando em consideração apenas o tipo de construção, se pressupõe uma relação direta entre a proximidade da fundação e do solo e a concentração dos contaminantes nos pontos de exposição. Isso porque há uma relação entre essa distância e a advecção (maior ou menor probabilidade de fissuras e interfaces piso-muro) e difusão (maior ou menor proximidade com a fonte). No entanto, o tipo de fundação por si só não determina a capacidade de intrusão de vapores, pois dependerá também dos processos construtivos. Por exemplo, em climas frios e secos, determinados países optam pelas fundações tipo entrepiso (crawlspace) com pouco espaço interno e uma rigorosa vedação, para evitar congelamento de dutos e otimização de calor. Porém, essa estrutura permite o acúmulo dos gases, mesmo com a aplicação de membranas supostamente impermeáveis no solo, por permitirem a passagem dos contaminantes por difusão.
Figura 6. Intrusão de vapor em ambientes internos para três cenários de construção de acordo com API (2005)
 4.1.5 Vias de exposição e Receptores
4.1.5 Vias de exposição e Receptores
As vias de exposição e os receptores serão tratados com detalhes no Capítulo 8 (Avaliação de Risco). Neste item são abordados parte dos conceitos, por estarem relacionados com a determinação da concentração final das substâncias químicas de interesse (SQI) em ambientes internos, como consequência da intrusão de vapores.
Como observado no modelo conceitual, a única via de exposição ou via de entrada dos contaminantes nos receptores é pela inalação de vapores. No entanto, os receptores têm características diferentes (como massa corpórea, frequência de exposição, taxa de contato etc.) inclusive de exposição. Os dados específicos associados à concentração final de cada SQI disponível para o receptor conformam a dose de ingresso, que é o parâmetro que, junto com o fator de risco (RF) e a dose de referência (RfD) permitem a quantificação do Risco à Saúde Humana do tipo carcinogênico ou não carcinogênico, respectivamente.
A quantificação da concentração final que receberão os receptores por meio da inalação (via de exposição) poderá ser obtida com auxílio de modelos analíticos, numéricos ou por medição direta por meio de amostragem de vapores.
|
Modelagem analítica A avaliação de risco à saúde humana, dependendo do modelo conceitual, requer uma grande quantidade de dados e, em alguns casos, cálculos complexos, comumente realizados com auxílio de softwares e planilhas de cálculo. Quando se trata de estimativas relacionadas com o mecanismo de transporte de gases por volatilização, a maioria desses programas (RISC, RBCA Tool Kit, SADA, CETESB entre outros) emprega o modelo de Johnson e Ettinger (1991), que embora tenha limitações (fonte infinita de contaminação; mistura do ar uniforme; não leva em consideração caminhos preferenciais nem biodegradação; considera distribuição do gás e taxas de ventilação uniformes; a entrada do gás é apenas por rachaduras da laje e juntas da laje-parede; análise unidimensional) é ainda um dos mais usados mundialmente. A expressão que relaciona a concentração no interior da edificação ou em ambiente interno (Cai) e concentração na fonte (Cf) é dada por:
No entanto, a fonte pode estar localizada tanto na água subterrânea como no solo. Para esses casos, a estimativa da concentração de vapor pode ser obtida pelas equações (12) e (13), respectivamente:
Onde: Cf é a concentração na fonte [M L-3], seja proveniente da concentração no solo Cs [-] ou da água subterrânea Cw [M L-3]. Cabe ressaltar que Cs não abrange a fase residual e Cw é menor que a solubilidade. rb é a massa específica do solo seco [M L-3] e Kd, o coeficiente de partição solo-água [L3 M-1]. Adicionalmente, considerando a hipótese de que a transferência de massa ocorre em estado estacionário (Johnson e Ettinger, 1991), a redução da concentração alcançada a partir da fonte até o ambiente interno pode ser estimada usando o coeficiente de atenuação (. O fator de atenuação da concentração de determinado contaminante leva em consideração diversos parâmetros, como: o transporte convectivo-difusivo; o coeficiente global de difusão efetiva da fase vapor através de rachaduras e juntas, cuja magnitude pode ser aproximada ao coeficiente de difusão efetiva da camada de solo em contato com a fundação; a espessura da fundação pertencente ao ambiente interno; a vazão de ventilação do ar no interior; as dimensões do ambiente interno, e a renovação do ar no ambiente interno. |


5. Reações
As concentrações de contaminantes em meio subterrâneo podem ser influenciadas por diferentes tipos de reações, que podem resultar na transformação ou degradação do contaminante. As reações resultam na redução de massa do contaminante original, e podem gerar substâncias inertes ou de menor risco, mas também podem gerar a formação de substâncias mais tóxicas. Diferentes tipos de reações podem ocorrer, destacando-se as reações químicas abióticas (sem o auxílio de microrganismos) e reações bióticas (mediadas por microrganismos).
As reações bióticas também são chamadas de biodegradação, e ocorrem de forma natural através de microrganismos que estão presentes na maioria dos solos. Os contaminantes orgânicos podem participar dessas reações de diversas formas. Em alguns casos, os contaminantes podem ser a fonte de carbono e energia para os microrganismos. Em outros casos, o contaminante é cometabolizado junto com a degradação de outro substrato primário. Ainda, o contaminante pode ser usado como aceptor de elétrons. A biodegradação é um importante processo de abatimento de massa de contaminantes, e por isso é explorado nas Medidas de Remediação por Tratamento (Seção 15.2) por meio das técnicas de biorremediação, incluindo a atenuação natural monitorada, método em que a biodegradação ocorre naturalmente sem intervenções além do monitoramento das condições do meio; e pelo bioestímulo e bioaumento, técnicas que induzem maiores taxas de biodegradação por meio da adição de receptores ou doadores de elétrons, nutrientes, controle das condições físico-químicas ou adição de culturas de microrganismos.
Diferentes tipos de reações abióticas podem ocorrer no meio subterrâneo, como hidrólise, reações de oxi-redução, dehalogenação, eliminação, precipitação e outras. A ocorrência dessas reações é dependente das propriedades da solução e da fase sólida, como temperatura, pH, potencial de oxirredução, presença de outros compostos químicos e composição química da fase sólida. Por exemplo, alguns minerais de ferro e manganês, e alguns grupos funcionais da matéria orgânica podem propiciar a ocorrência de redução química abiótica. Essas reações abióticas em subsuperfície podem ser utilizadas como Medidas de Remediação por Tratamento (Seção 15.2) através da Redução Química in Situ (ISCR) e Oxidação Química in Situ (ISCO).
Alguns exemplos de reações importantes são descritos na sequência.
5.1 Biodegradação de hidrocarbonetos
A redução da massa de compostos orgânicos é mediada pelo metabolismo microbiano. As rotas metabólicas envolvidas na mineralização de hidrocarbonetos são dependentes da disponibilidade de aceptores de elétrons. Quando o oxigênio está disponível em concentrações suficientemente elevadas, existem condições propícias para a biodegradação aeróbica, descrito abaixo para o tolueno:
![]()
Na ausência do oxigênio, a biodegradação de hidrocarbonetos vai se suceder anaerobicamente a partir das reações de redução do nitrato, redução do Fe(III), redução do sulfato e metanogênese (que formam metano); apresentadas nas Equações 15 a 18, respectivamente. Essas reações ocorrerão prioritariamente a partir de reações termodinamicamente mais favoráveis (maior energia livre de Gibbs). Quando os aceptores de elétrons não estão mais disponíveis no aquífero, as reações de metanogênese (Equação 18) são dominantes.
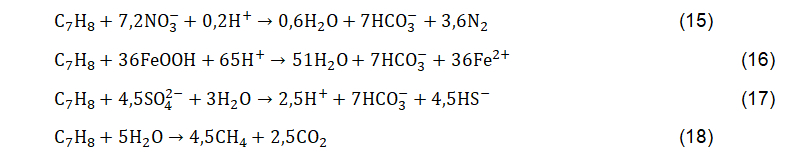
Nas reações de biodegradação anaeróbica, acima apresentadas, é possível verificar que a biodegradação de hidrocarbonetos resulta na produção de CO2, N2, Fe(II), HS– e CH4. O Fe(II) e o CH4, produzidos respectivamente por redução do Fe(III) e metanogênese, representam indicadores comumente empregados para comprovação da ocorrência de biodegradação. Cabe destacar que embora a metanogênese seja tradicionalmente classificada como uma reação com energia livre amplamente reduzida, as relações de sintrofia podem tornar essa reação eficiente do ponto de vista termodinâmico. Concentrações elevadas dessas espécies são correlacionáveis com concentrações elevadas de compostos BTEX na água subterrânea.
5.2 Degradação de compostos halogenados
A desalogenação redutiva é um modo importante de degradação de vários compostos, incluindo pesticidas organoclorados, solventes alquílicos e haletos de arila, podendo ocorrer a partir de rotas metabólicas abióticas ou bióticas. A ação metabólica de alguns microrganismos possui a capacidade de remover halogênios (Cl-, Br-, F- e I-) de compostos orgânicos halogenados em ambientes anóxicos. A desalogenação pode ocorrer por meio de uma variedade de reações, incluindo oxidação, redução ou hidrólise.
O processo de remoção de um halogênio de um composto orgânico halogenado por uma reação redutiva é conhecido como desalogenação redutiva. A reação de desalogenação redutiva com hidrogênio como o doador de elétrons é altamente exergônica, sendo que a maioria dos organohaletos são termodinamicamente favoráveis como aceptores de elétrons em condições anaeróbias (Judger et al. 2016, Holliger et al., 1998). Nas reações redutivas, os elétrons são transferidos para a ligação carbono-halogênio, diminuindo assim o estado de oxidação do composto original, podendo ser classificados em dois processos distintos: hidrogenólise e dihaloeliminação.
Reação de Hidrogenólise
A hidrogenólise também ocorre para compostos aromáticos clorados, incluindo benzenos clorados, bifenilos policlorados (PCBs), dioxinas cloradas (por exemplo, 2,3,7,8-tetraclorodibenzo-p-dioxina ou TCDD), furanos e fenóis policlorados (por exemplo, pentaclorofenol). Para esses compostos, as vias de degradação são complexas, pois a redução ocorre por meio de vários isômeros, dependendo da posição no anel em que cada átomo de cloro específico é removido e, por esse motivo, as reações não serão apresentadas.
No caso dos etanos e metanos clorados, a hidrogenólise de compostos aromáticos clorados raramente é completa, e a taxa de redução frequentemente diminui com a diminuição do número de ligações cloro-carbono. O processo normalmente ocorre por meio de um processo respiratório conhecido como respiração organohalide. A hidrogenólise de etenos clorados, tal como o tetracloroeteno (PCE), prossegue em duas etapas por meio de tricloroeteno (TCE), dicloroetenos (cis- ou trans-1,2-DCE), cloreto de vinila e eteno, conforme representado na Figura 7. A presença de 1,1-DCE no meio ambiente está mais tipicamente associada à contaminação prévia por 1,1,1-tricloroetano, que sofre diversos tipos de transformação, incluindo o processo abiótico de desidrohalogenação em 1,1-DCE.
Figura 7 Decaimento de PCE para TCE, cis- ou trans-1,2-DCE, cloreto de vinila e eteno a partir de reações de hidrogenólise
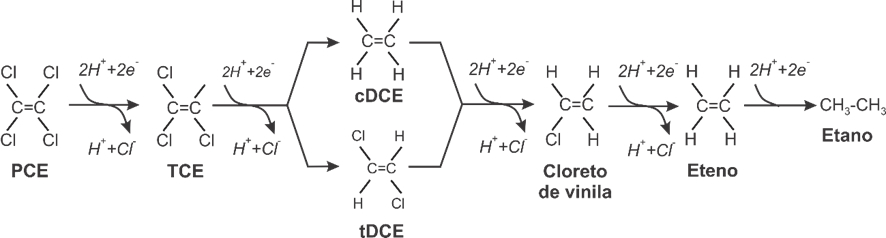
O estado de oxidação do carbono em um composto orgânico varia de -4 (CH4) a +4 (CO2), verificando-se a tendência de a oxidação ocorrer mais intensamente nos compostos mais reduzidos. O estado de oxidação do carbono em PCE é +4. Com cada etapa de redução sucessiva (por meio de uma entrada de 2e-), o estado de oxidação do carbono diminui em 2, de modo que o carbono em TCE tem um estado de oxidação de +2, DCE tem 0, o cloreto de vinila tem -2 e eteno tem -4. Para essa categoria de contaminantes, a etapa final é crítica do ponto de vista ambiental, uma vez que o cloreto de vinila possui propriedades carcinogênicas e elevada volatilidade, enquanto o eteno e o etano não apresentam riscos à saúde humana.
Como os etenos clorados, os etanos clorados são reduzidos por hidrogenólise. Um dos compostos mais amplamente avaliados é o 1,1,1-tricloroetano, que sofre redução sequencial para 1,1-dicloroetano e cloroetano. Embora seja possível uma redução adicional em etano, é uma reação muito mais lenta e o cloroetano é tipicamente considerado como o produto terminal. Esse exemplo serve para ilustrar que a hidrogenólise nem sempre resulta em descloração completa. Outros etanos clorados comumente encontrados sofrem hidrogenólise, incluindo redução de 1,2-dicloroetano em cloroetano e 1,2-dicloropropano em 1- ou 2-cloropropano.
Os metanos clorados, como o tetracloreto de carbono (tetraclorometano) sofrem hidrogenólise para clorofórmio (triclorometano) e depois cloreto de metileno (diclorometano). Essas reações também são catalisadas pela redução do ferro, que pode ser gerado por bactérias redutoras de ferro.
Reação de Dihaloeliminação
A redução dos compostos orgânicos halogenados pode ser conduzida pela dihaloeliminação, uma reação descrita por Sulita et al. (1982) e menos abundante do que a hidrogenólise. Essa reação consiste na remoção de átomos de halógenos de carbono adjacentes, resultando na formação de uma ligação dupla de carbono e liberação de duas moléculas de halógenos. Um exemplo de reação de dihaloeliminação a partir da redução do 1,1,2-tricloroetano para cloreto de vinila é expresso na equação abaixo.
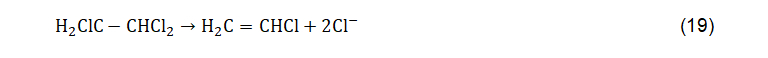
6. Referências
API – AMERICAN PETROLEUM INSTITUTE. A Practical Strategy for Assessing the Subsurface Vapor-to-Indoor Air Migration Pathway at Petroleum Hydrocarbon Sites: Collecting and Interpreting Soil Gas Samples from Vadose Zone. Publication 4741. Washington D. C: Regulatory Analysis and Scientific Affairs. 2005.
CETESB. Companhia Ambiental do Estado de São Paulo. Planilhas para avaliação de risco em áreas contaminadas sob investigação. 2013. Disponível em: https://cetesbhomolog.sp.gov.br/areas-contaminadas/documentacao/planilhas-para-avaliacao/. Acesso em: 08 de dezembro de 2020.
CETESB. Cia. de Tecnologia e Saneamento Ambiental do Estado de São Paulo. Relação de Áreas Contaminadas e Reabilitadas no Estado de São Paulo, Texto Explicativo, São Paulo, 389p. 2018. Disponível em: https://cetesbhomolog.sp.gov.br/areas-contaminadas/wp-content/uploads/sites/17/2018/01/Texto-explicativo.pdf. Acesso em: 01 dez. 2020.
CL:AIRE. An illustrated handbook of LNAPL transport and fate in the subsurface. CL:AIRE, London. ISBN 978-1-905046-24-9. 2014.
DOMENICO, P.A.; SCHWARTZ, F.W. Physical and Chemical Hydrogeology. 2nd Edition. John Wiley & Sons: New York. 1998.
DUNNIVANT, F. M.; ANDERS, E. A basic introduction to pollutant fate and transport: an integrated approach with chemistry, modeling, risk assessment, and environmental legislation. Wiley-Interscience: Hoboken. 480 p. 2006.
FETTER, C.W. Applied Hydrogeology. Prentice Hall: New Jersey, 4a edição. 2001.
FITTS, C.R. Águas Subterrâneas. Elsevier: Rio de Janeiro. 2ª edição. 2015.
GIEG, Lisa M.; FOWLER, S. Jane; BERDUGO-CLAVIJO, Carolina. Syntrophic biodegradation of hydrocarbon contaminants. Current opinion in biotechnology, v. 27, p. 21-29, 2014.
HOLLIGER, Christof; WOHLFARTH, Gert; DIEKERT, Gabriele. Reductive dechlorination in the energy metabolism of anaerobic bacteria. FEMS Microbiology Reviews, v. 22, n. 5, p. 383-398, 1998.
ITRC (2014). Interstate Technology and Regulatory Council. Petroleum Vapor Intrusion: Fundamentals of Screening, Investigation, and Management. Washington, D.C.: Interstate Technology & Regulatory Council, Vapor Intrusion Team. Disponível em: www.itrcweb.org/PetroleumVI-Guidance/Content/Resources/PVIPDF.pdf. Acesso em: 10 nov.2020
JOHNSON, P. C.; ETTINGER, R. A. (1991). Heuristic Model for Predicting the Intrusion Rate of Contaminant Vapors into Buildings. Environmental Science & Technology, v. 25, n. 8, p.1445-1452.
JUGDER, Bat-Erdene et al. Organohalide respiring bacteria and reductive dehalogenases: key tools in organohalide bioremediation. Frontiers in microbiology, v. 7, p. 249, 2016.
LENHARD, R. J.; PARKER, J. C. Estimation of free hydrocarbon volume from fluid levels in monitoring wells. Ground water, v. 28, n. 1, p. 57–67, 1990.
MAYER, A. S.; HASSANIZADEH, S. M. Soil and Groundwater Contamination: Nonaqueous Phase Liquids—Principles and Observations. Washington, D. C.: American Geophysical Union, 2005.
MC WORTHER D.B. Flux equation for gas diffusion in porous media. The Groundwater Project. Guelph, Ontario, Canada. 2021. Disponível em: <http://gw-project.org >. Acesso em: 10 ago 2021.
MILLINGTON R.J.; QUIRK. J.P. Permeability of Porous Media, Transactions of the Faraday Society, Vol. 57, pg. 1200-1207, 1961.
MOHN, William W.; TIEDJE, James M. Microbial reductive dehalogenation. Microbiology and Molecular Biology Reviews, v. 56, n. 3, p. 482-507, 1992.
NAZAROFF, W.W. Predicting the rate of 222Rn entry from soil into the basement of a dwelling due to pressure-driven air flow. Radiation Protection Dosimetry, vol. 24, pg. 199-202, 1988.
NEILSEN. D.R.; JACKSON, R.D.; CARY, J.W.: EVANS, D.D. Soil Water. American Society of Agronomy, Madison, Wisconsin. 175 pp. 1972.
NJDEP (2005). New Jersey Department of Environmental Protection. Field Sampling Procedures Manual. Site Remediation and Waste Management Program. Disponível em: www.nj.gov/dep/srp/guidance/fspm/. Acesso em 19 out.2020
SHARMA, P. K.; MUSKAN MAYANK; OJHA, C.S.P.; SHUKLA, S. K. A Review on Groundwater Contaminant Transport and Remediation. ISH Journal of Hydraulic Engineering, v. 26, n. 1, p. 112–121. 2020.
SUFLITA, Joseph M. et al. Dehalogenation: a novel pathway for the anaerobic biodegradation of haloaromatic compounds. Science, v. 218, n. 4577, p. 1115-1117, 1982.
TOMLINSON, D. W.; RIVETT, M. O.; WEALTHALL, G. P.; SWEENEY, R. E. H. Understanding complex LNAPL sites: Illustrated handbook of LNAPL transport and fate in the subsurface. Journal of Environmental Management, v. 204, p. 748–756, 2017.
USEPA (2001). United States Environmental Protection Agency. OSWER: Fact Sheet: Correcting the Henry’s Law Constant for Soil Temperature. Disponível em: https://www.epa.gov/risk/usepa-oswer-fact-sheet-correcting-henrys-law-constant-soil-temperature. Accesso em 22/08/2021.
USEPA (2002). United States Environmental Protection Agency. Draft Guidance for Evaluating the Vapor Intrusion to Indoor Air Pathway from Groundwater and Soils. Washington, D.C.: Office of Solid Waste and Emergency Response. Disponível em: www.epa.gov/correctiveaction/eis/vapor/complete.pdf. Accesso em 01/02/2015.
USEPA. United States Environmental Protection Agency. User’s Guide for Evaluating Subsurface Vapor Intrusion into Buildings. Revised. Office of Emergency and Remedial Response. 2004. Disponível em: www.epa.gov/oswer/riskassessment/airmodel/pdf/2004_0222_3phase_users_guide.pdf. Acesso em 15/10/2020
USEPA. United States Environmental Protection Agency. Assessment of Mitigation Systems on VI: Temporal Trends, Attenuation Factors, and Contaminant Migration Routes under Mitigated and Non-mitigated Conditions, EPA/600/R-13/241, Washington, D.C., USA. 2015.
WIEDEMEIER, Todd H. et al. Natural attenuation of fuels and chlorinated solvents in the subsurface. John Wiley & Sons, 1999.

